C(sp3)−H键的官能化反应一直被视为有机化学的圣杯。此类反应可以将廉价易得的烷烃资源转化成高附加值的化学产品,对石油化工行业产业升级具有重要意义。由于C(sp3)−H键能大、极性小且广泛存在于各类有机化合物中,其反应活性低且选择性难以控制。另一方面,相关反应的中间体众多且难以分离与检测,这使得C(sp3)−H键官能化反应机理方面的研究也面临重大挑战。
近期,bat365官网登录入口/理论化学研究所青年教师钟荣林博士与京都大学触媒∙电池元素战略研究基地高级研究员Sakaki Shigeyoshi(榊茂好)荣誉教授合作,在此领域取得重要进展,连续在J. Am. Chem. Soc上发表两篇长文,深入地阐释了控制底物活性、区域选择性、化学选择性和催化剂活性的关键因素,并以此为基础对相关催化剂进行了精准的结构设计。
2019年,钟荣林博士作为第一作者在J. Am. Chem. Soc上发表了该类反应机理的重要进展[1]。研究发现,C(sp3)原子轨道能是引起电子效应且控制C(sp3)−H键反应活性和区域选择性的一个关键因素(图1)。他们探索了配体对催化剂活性的影响规律,为后续相关研究奠定了基础。该项工作发表以来,已被多个实验研究团队关注并引用。加州大学伯克利分校的Hartwig教授最近在Science上发表的相关研究论文[2]中,三处引用并积极评价了钟荣林博士等在该类反应机理方面的贡献。

图1 THF的βC(sp3)−H键区域选择性反应及其内在原因。
近期,钟荣林博士继续在此领域深入探索,将对机理的研究拓展到在实验上更难实现的甲烷硼化反应,并作为第一作者和通讯作者,以bat365官网登录入口理论化学研究所为第一通讯单位,在J. Am. Chem. Soc上发表了有关机理研究与催化剂精准设计的工作[3]。该工作主要发现了Ru催化的甲烷硼化过程中的两个中间体都包含较强的H…Bpin成键相互作用,它们中Ru的氧化态是一个介于Ru(IV)和Ru(VI)的特殊中间态。在对催化剂的活性和化学选择性深入理解之后,他们进一步提出了一个二者兼顾的配体设计策略,改善铱催化剂的性能:利用具有非对称结构且强供电子能力的氮杂环卡宾-吡啶配体来增强铱配合物对甲烷的催化活性,同时在合适的位置引入特定的位阻基团来压制溶剂分子的反应并提高催化剂对甲烷硼化的化学选择性。通过对13种配体参与的反应进行详细理论计算,预测了可能表现出更高活性且更高化学选择性的相关催化剂结构,实现了对催化剂结构的精准设计(图2)。
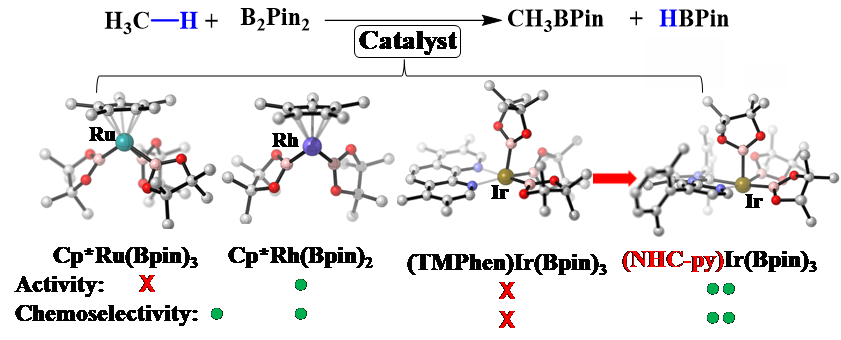
图2 甲烷硼化反应,相关活性物种的结构及其催化活性与化学选择性。
上述理论研究工作以唐敖庆先生发展的配位场理论方法与福井谦一先生发展的前线分子轨道和IRC理论为基础,充分发挥理论与计算化学在研究催化反应机理和辅助催化剂设计方面的显著优势。一方面有助于加深相关领域研究人员对C(sp3)−H键活化机理的基础理解,另一方面也为催化剂的高效合成与反应条件优化提供了可行方案。高效实现甲烷等烷烃资源的转化将有力推动当前石油化工产业的变革,上述研究有望为将来发展相关产业合成技术奠定基础,促进烷烃资源的综合利用。
相关阅读:
[1]2019年论文链接:https://pubs.acs.org/doi/10.1021/jacs.9b01767。
[2]Hartwig教授的论文链接:https://science.sciencemag.org/content/368/6492/736。
[3]2020年论文链接:https://pubs.acs.org/doi/10.1021/jacs.0c07239。



 当前位置:
当前位置:

