近日,bat365官网登录入口、理论化学研究所李辉教授课题组在《Journal of Chemical Theory and Computation》上发表了题为” MLRNet: Combining the Physics-Motivated Potential Models with Neural Networks for Intermolecular Potential Energy Surface Construction”的研究工作。该工作讨论了一种“物理模型+神经网络”的理论框架,通过将神经网络嵌入物理势能函数的方式,同时结合神经网络和物理模型在势能拟合中的优势。基于该框架,作者提出了一种用于光谱精度分子间相互作用势拟合的神经网络模型,MLRNet模型。该模型突破了传统神经网络模型在相互作用长程和短程渐近区拟合效率低、外推误差大的问题,并展现出很高的拟合精度和高维拟合效率,为未来神经网络势能模型的发展提供了新思路。


图一:“物理模型+神经网络”框架和MLRNet模型的基本思路。
势能拟合是连接电子结构计算与分子动力学模拟之间的桥梁,一般需要优化给定势能模型的参数,从而使该势能模型尽可能真实地反映电子结构计算的能量。神经网络作为平滑函数的“万能近似器”,在势能拟合领域备受关注。如今已经有许多神经网络势能模型被应用在不少小分子体系反应动力学、分子-表面相互作用、以及凝聚相体系的分子动力学模拟等工作中。然而作为“数据驱动”的模型,神经网络在缺乏足够数据或可靠对称性约束时很容易出现非物理的外推。对于拥有明确物理极限的长程渐进区,神经网络直接拟合的效率非常低,同时外推结果也通常不具有物理意义。如何发挥神经网络技术在函数拟合方面的优势,同时严格保证体系已知物理特性,是神经网络势能模型发展的一个前沿课题。
李辉课题组的一些工作致力于用“物理驱动”的mdMLR模型拟合分子间相互作用势(Mol. Phys. 116, 843 (2018)、J. Chem. Phys. 148, 124302 (2018)、J. Chem. Phys. 153, 54303 (2020))。该模型具有良好的短程和长程外推能力,在一些范德华体系的高精度光谱研究和低温超流模拟中发挥着重要作用。虽然mdMLR在6维以下的势能面拟合具有很高的精度,但mdMLR模型参数的线性展开形式是直积求和的,严重降低了该模型在高维度势能拟合的效率。
基于上述问题,李辉课题组将神经网络模型和mdMLR模型有机结合,发展了MLNet模型,从而充分发挥两者在势能拟合上的优势。该工作中的MLRNet模型采用独立的子网络表示mdMLR函数中的物理参数。考虑到神经网络通常不具备置换对称性和mdMLR模型对输入变量的需求,该工作采用了分子质心间距和该间距无关的置换不变多项式作为模型的输入,从而在保证了物理对称性的同时,满足mdMLR势能函数对参数的基本要求。
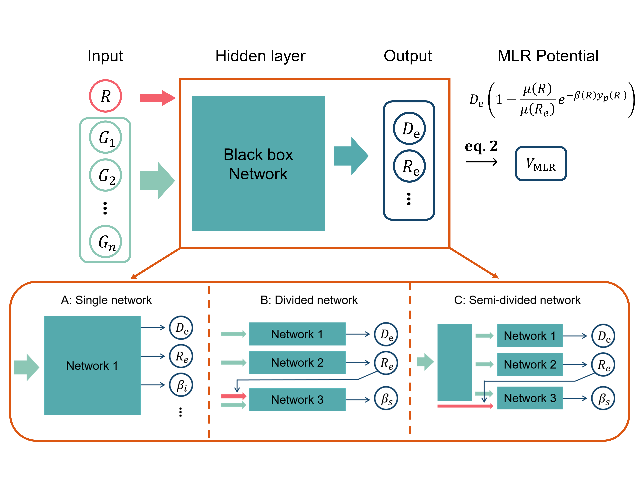
图二:MLRNet可以采用的神经网络结构。
该工作在CO2-He体系和H2O-Ar体系测试了MLRNet模型。MLRNet不仅表现出了很高的拟合精度,拟合误差相比mdMLR模型结果降低了数倍(MLRNet: 0.005 cm−1 vs mdMLR: 0.032 cm−1),而且具有良好的长程和短程外推精度。此外,在H2O-Ar体系的测试中,MLRNet展现了很高的模型效率:仅需1509个参数即可达到3501个参数的置换不变多项式神经网络模型(PIP-NN)的拟合精度,并且与5维mdMLR拟合所需的参数接近(1625)。更少的模型参数意味着MLRNet可以节省动力学模拟中势能调用的成本,进而减少总模拟时长。
该工作相关的研究成果已发表在Journal of Chemical Theory and Computation杂志上,文章第一作者为吉林大学博士生李由,通讯作者为吉林大学李辉教授。该工作得到了国家自然科学基金的支持。
文章详情:You Li, Yu Zhai, Hui Li, “MLRNet: Combining physics-motivated potential models with neural networks for intermolecular potential energy surface construction”, Journal of Chemical Theory and Computation, 19, 1421 (2023).
全文链接:https://pubs.acs.org/doi/10.1021/acs.jctc.2c01049
课题组网页:http://huiligroup.org/zh/index.html



 当前位置:
当前位置:

